


Генетичні захворюванняСиндром Блума Синдром Блума (СБ), (англ. Bloom syndrome), також відомий як синдром Блума-Торре-Махачека - це рідкісний аутосомно-рецесивний хромосомний розлад, який характеризується високою частотою розривів і перебудов в хромосомах ураженої людини. Цей синдром був вперше описаний лікарем дерматологом Девідом Блумом (Dr. David Bloom) у 1954 році. Ознаки і симптоми.
Особи, які хворі на СБ, як правило не високого росту, з характерними висипами на шкірі, які виникаютьмайже одразу після першого впливу сонячних променів. Ці висипи, як правило виникають на щоках (викликаючи почервоніння) та мають форму метелика. Але висипи можуть виникати не лише на обличчі, але й на інших частинах тіла, на яких потрапляло сонячне проміння (спина, руки, тощо).
Інші клінічні ознаки:
Ускладнення, які виникають у результаті дії захворювання, можуть включати в себе хронічні хвороби легень, цукровий діабет, виникнення труднощів під час навчання. У деяких осіб, виникає розумова відсталість. Найбільш небезпечним ускладненням можна назвати підвищення ймовірності виникнення пухлинних захворювань.
 Зв'язок із виникненням раку Внаслідок великої кількості мутацій, які відбуваються при захворюванні на синдром Блума, в уражених осібсуттєво підвищується ризик захворіти на рак. Схильність до онкологічних захворювань характеризується:
1) широким спектром прояву хвороб, який включає лейкози, лімфоми і карциноми;
2) раннім віком виникнення пухлинних захворювань, порівно до решти населення;
3) складністю та важкістю перебігу.
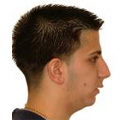
Рис. 1. Мікрогнатія
У осіб з синдромом Блума рак може виникнути у будь-якому віці. Середній вік хворих – 25 років. Патофізіологія Мутації в гені BLM, який являється складовою ДНК геліказ (хелікази), спричиняють виникнення синдрому Блума. ДНК гелікази (хелікази) - це ферменти, які розкручують дволанцюгову спіраль ДНК. Процес «розкручування» складається з великої кількості процесів, таких як синтез копій ДНК, транскрипція РНК, репарація ДНК і т.д. Коли клітина готується до поділу, то хромосоми дублюються таким чином, що кожна новоутворена
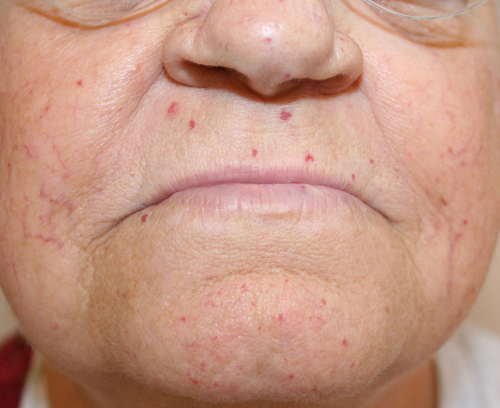 структура отримує повний набір хромосом. Процес подвоєння називається - реплікацією ДНК. Помилки, допущені під час реплікації ДНК можуть призвести до мутації. Білок BLM відіграє важливу роль у підтримці стабільності ДНК під час процесу реплікації. Мутації гена BLM, які пов'язані з синдромом Блума дезактивують білок, (який кодується геном BLM) і входить до складу ДНК геліказ, або ж порушують процес експресії (утворення білка). Іншими словами не відбувається експресія цього гену. Відсутність BLM білка або низька його активність призводить до збільшення кількості мутацій, саме тому молекулярні механізми, з допомогою яких BLM підтримує стабільність хромосом, досі залишаються предметом для багатьох структура отримує повний набір хромосом. Процес подвоєння називається - реплікацією ДНК. Помилки, допущені під час реплікації ДНК можуть призвести до мутації. Білок BLM відіграє важливу роль у підтримці стабільності ДНК під час процесу реплікації. Мутації гена BLM, які пов'язані з синдромом Блума дезактивують білок, (який кодується геном BLM) і входить до складу ДНК геліказ, або ж порушують процес експресії (утворення білка). Іншими словами не відбувається експресія цього гену. Відсутність BLM білка або низька його активність призводить до збільшення кількості мутацій, саме тому молекулярні механізми, з допомогою яких BLM підтримує стабільність хромосом, досі залишаються предметом для багатьох Рис. 2. Телеангіектазія досліджень. У осіб, з СБ мають місце великі кількості обмінів між гомологічними хромосомами або сестринськими хроматидами (дві молекули ДНК, які утворюються в процесі реплікації ДНК),суттєво зростає кількість мутацій. Крім того, на багато частіше (по відношенню до осіб, які не мають синдрому Блума) трапляється пошкодження хромосом та їхня перебудова. Сьогодні вчені працюють над тим, що встановити (чи спростувати) наявність прямого зв’язку між молекулярними процесами, в яких бере участь ген BLM та зміною хромосом. Крім того, зв'язок між молекулярними дефектами в клітинах осіб з СБ, хромосомними мутаціями, які накопичуються в соматичних клітинах (усіх клітинах тіла, окрім статевих) та багатьма клінічними ознаками синдрому Блума також є одним із напрямків клінічних досліджень.
Синдром Блума успадковується по аутосомно-рецесивному типу. Тобто, обоє батьків мають бути носіями для того, щоб їхня дитина захворіла. Носіями захворювання серед євреїв Ашкеназі, що переважно походять із Східної Європи, є 1людина на 100 осіб. Якщо обидва батьки є носіями СБ, то у 25% випадків, дитина може бути хворою.
Тим членам сімей, які можуть бути носіями захворювання рекомендується звернутися з проханням про генетичну консультацію або ж пройти генетичне тестування. Для носіїв-батьків, які планують народжувати дитину варто провести пренатальну діагностику за допомогою цитогенетичних та молекулярних методів. Також можливо здійснити молекулярний аналіз ДНК для визначення наявності мутацій. Інші мутації при синдромі Блума Вперше дві міссенс мутації ферменту піруват кінази М2 (H391Y і K422R), виявили в клітинах хворих на синдром Блума, як потім підтвердилося вони підвищують схильність до виникнення раку. Результати показують, що незважаючи на наявність мутацій в ділянці домену міжсубодиничного контакту, мутантні білки K422R і H391Y зберігають свою гомотетраметричну структуру, що робить їх схожими по структурі на звичайні білки, але їхня активність суттєво зменшується на 75 і 20% відповідно. Цікаво, що H391Y показав 6-кратне збільшення спорідненості зі своїм субстратом фосфоенолпіруватом і поводив себе як неалостеричний білок із порушеним з’єднанням (із субстратом). Проте, K422R втрачає свою спорідненість із фосфоенолпіруватом. На відміну від K422R, H391Y проявляє поліпшені теплові характеристики і стабільність в певних діапазонах значень рН, крім того спостерігається менший вплив алостеричних інгібіторів Phe, і стійкість під час структурних змін при зв'язуванні активатора (фруктозо 1,6-бісфосфату) і інгібітора (Phe). Обидва мутанти показали невелике зрушення в оптимумі рН від 7,4 до 7,0. Спільна експресія гомотетрамерів дикого типу та мутанта PKM2 в клітинному середовищі, яка виникає в результаті взаємодії між ними на рівні мономерів, була пізніше підтверджена під час клінічних експериментів. Крос-мономерна взаємодія суттєво змінює олігомерний стан PKM2, через посилення димеризації і гетеротетрамерізації Ці особливості, як передбачається, відіграють важливу роль під час пухлинної прогресії. PKM2 (M2-PK); потенціал багатофункціонального білка Висловлено припущення, що аберантний білок PKM2 може бути основною причиною виникнення інших симптомів, характерних для синдрому Блума.
<<<
Алькаптонурія
Гістидинемія
Гомоцистинурія
Полікістозна хвороба нирок
Хвороба Вільсона – Коновалова
Cиндром Марфана (Хвороба Марфана)
X-зчеплений іхтіоз
Анемія Фанконі
Аргініносукцинатна ацидурія
Бета-таласемія
Бічний аміотрофічний склероз (частина друга)
Бічний аміотрофічний склероз (частина перша)
Галактоземія
Гемоглобін Е
Гемоглобін С
Гемохроматоз
Глікогенози
Дальтонізм (частина друга)
Дальтонізм (частина перша)
Дефіцит 3-гідкрокси-метил-глутарил КоА ліази
Дефіцит 3-метилкротоніл-коензим А карбоксилази
Дефіцит альфа 1 антитриптисину
Дефіцит бета-кетотіолази
Дефіцит біотинідази
Дефіцит дигідропіримідин дегідрогенази
Дефіцит довголанцюгової ацил-коензим А дегідрогенази
Дефіцит коротколанцюгової ацил-коензим А дегідрогенази
Дефіцит метилентетрагідрофолат редуктази
Дефіцит прекалікреїну
Дефіцит синтетази голокарбоксилази
Дефіцит фактора ХІ
Ентеропатичний акродерматит
Іміногліцинурія
М'язова дистрофія Дюшена
Муковісцидоз
Нейрофіброматоз
Органічні ацидемії
Первинний системний дефіцит карнітину
Перманентний неонатальний цукровий діабет
Перонеальна дистрофія Шарко-Марі -Тута (частина друга)
Перонеальна дистрофія Шарко-Марі-Тута (частина перша)
Пропіонова ацидурія
Псевдополідистрофія Гурлера
Серповидно-клітинна анемія (частина друга)
Серповидно-клітинна анемія (частина перша)
Синдром Ангельмана
Синдром Бартера
Синдром Блума
Синдром Дабіна Джонсона
Синдром Дауна (частина друга)
Синдром Дауна (частина перша)
Синдром Едвардса
Синдром Клайнфельтера-Рейфенштейна-Олбрайта
Синдром котячого крику (5р)
Синдром Кріглера Найара
Синдром Леша-Наяна
Синдром Лойса-Дітца
Синдром Патау
Синдром Прадера-Віллі
Сімейна вегетативна дисфункція
Сімейна гіперхолестеринемія
Тирозинемія
Тирозинемія І типу
Тирозинемія ІI типу
Тирозинемія ІIІ типу
Фенілкетонурія (ФКУ)
Фруктоземія
Хвороба I клітин
Хвороба Гірке
Хвороба Дауна
Хвороба Канавана
Хвороба кленового сиропу
Хвороба Краббе
Хвороба Німана – Піка
Хвороба Тея-Сакса (друга частина)
Хвороба Тея-Сакса (перша частина)
Хвороба Фабрі
Хвороба фон Віллебранда
Хвороба Хантінгтона
Целіакія (частина друга)
Целіакія (частина перша)
Церебротендінальний ксантоматоз
|